Dhatri Badri's Home Page

Hi! My name is Dhatri Badri and I am a aspiring Bioinformatician.
Currently, I am a graduate student at Boston University getting my Masters in Bioinformatics. I graduated George Washington University in Cell and Molecular Biology minoring in Bioinformatics and STEM teaching.
View My LinkedIn Profile
View My Resume
View My Teaching Website
Characterizing vermicompost microbiome based on shotgun and amplicon sequecing
Project description: This project aimed to compare inferences from metagenomic and metataxonomic data to characterize the diversity of the bacterial and fungal communities associated with vermicomposting. Vermicomposting takes advantage of the synergistic effects of worms and microorganisms (bacteria and fungi) to decompose organic waste. Furthermore, worms can enrich the microbial communities of the resulting compost, enhancing subsequent production as a richer form of organic fertilizer, thus offering a sustainable alternative to chemicals while converting waste products into fertilizer.
1. How was data processed?
Microbial profiles were obtained by shotgun sequencing of genomes and through amplicon sequencing of 16S rRNA for bacteria and ITS for fungi.
Data from 16S and 18S sequencing were processed using the R package dada2 version 1.12. Then, the standard dada2 pipeline was applied to perform amplicon sequence variant (ASV) inference, merge paired reads, and identify chimeras. Taxonomic assignment for 16S was performed against the Silva v132 database using the dada2-formatted training files for taxonomy and species-level assignment. Taxonomic assignment for 18S was performed against the Silva v132 Eukaryotic 18S database.
Data from ITS sequencing were processed differently due to the variable size of the ITS region. Raw reads from ITS sequencing were first subject to adapter trimming by cutadapt version 2.3 which removed primer sequences due to read-through. Taxonomic assignment for ITS was performed against the UNITE version 18.11.2018 database.
For the resulting 16S, 18S, and ITS dada2-processed data, ASV sequences were aligned using MAFFT and used to build a tree with FastTree. The resulting data were imported into phyloseq for further analysis.
2. What kind of data was I dealing with?
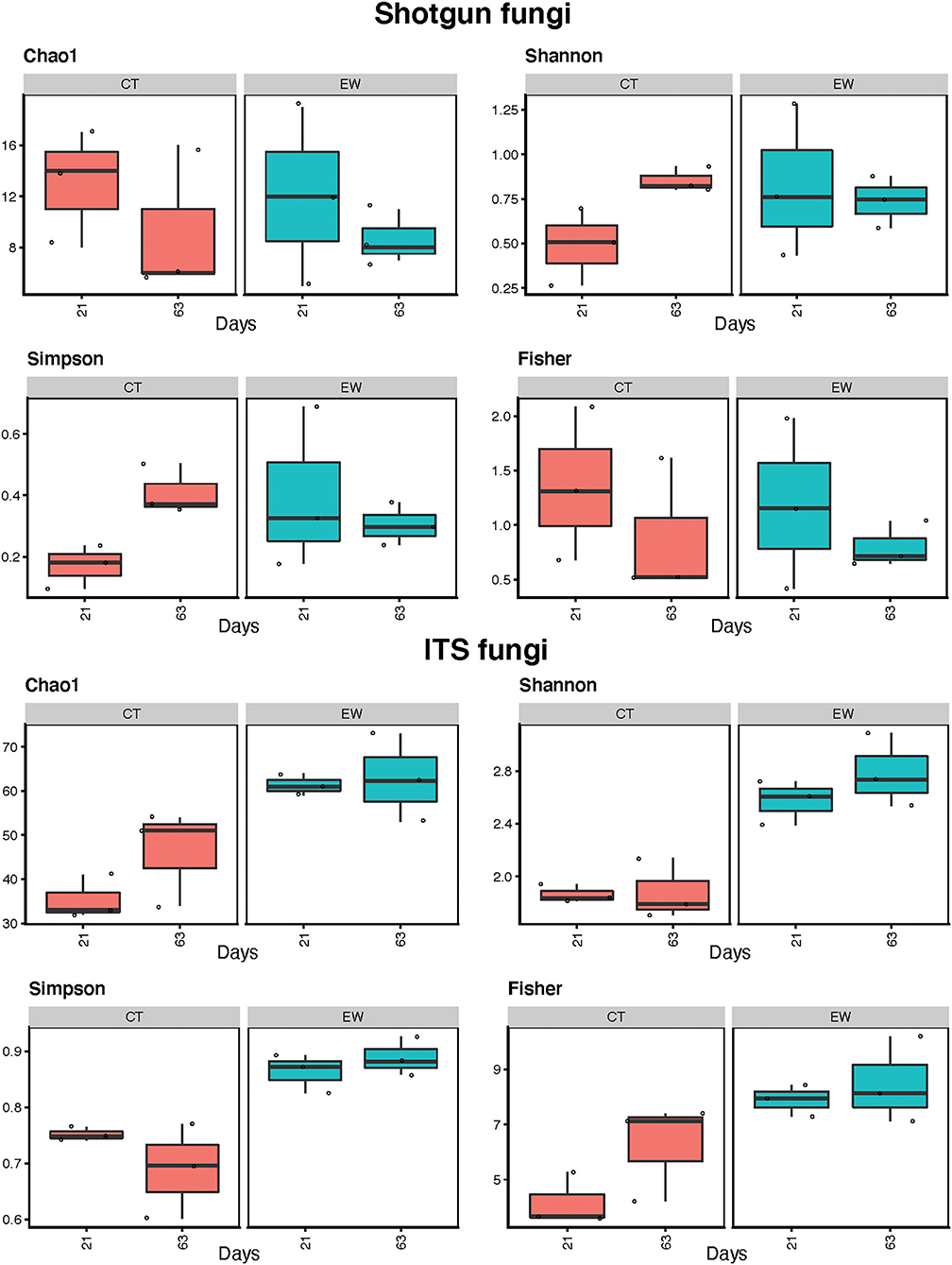
It is imperative to ask the right questions to elicit the the right responses. Overview of abundant taxa in each group—in phlya and class. Alpha and beta diversity indices.
Alpha diversity: Chao, Shannon, Simpson, Fisher
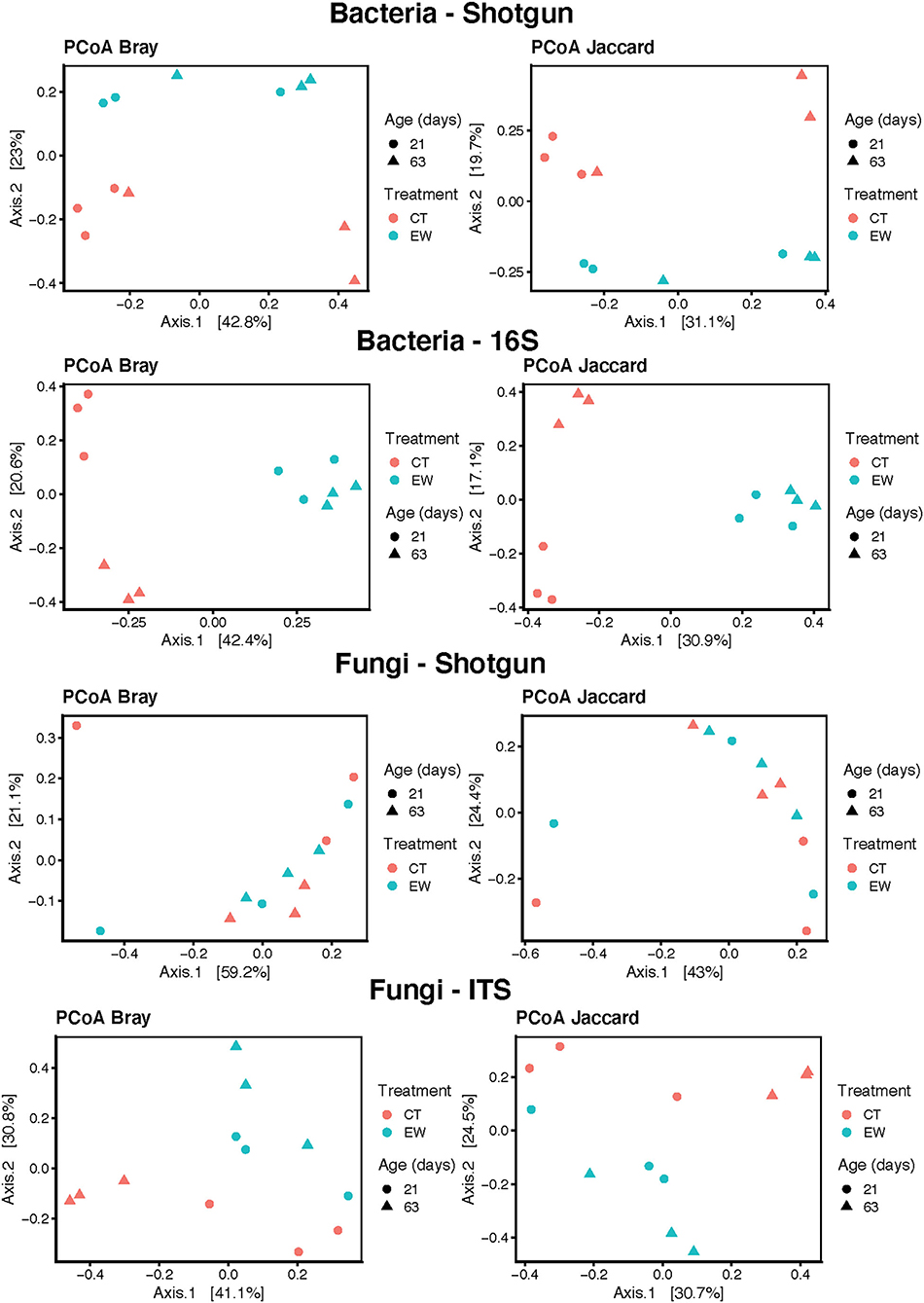
Beta diversity: Bray, Jaccard
Questions
1. What are the most abundant bacterial and fungal taxa in each group?
2. Does taxonomic composition vary the same way across groups?
3. Does alpha-diversity vary the same way across groups?
4. Does beta-diversity vary the same way across groups?
5. What are the most abundant bacterial functions in each group?
6. Do bacterial functional profiles vary the same way across groups?
3. How was data analyzed?
Unfortunately, I was unable to include all my analyses so I selected.